# 安装包
if (!requireNamespace("circlize", quietly = TRUE)) {
install.packages("circlize")
}
if (!requireNamespace("ComplexHeatmap", quietly = TRUE)) {
install_github("jokergoo/ComplexHeatmap")
}
if (!requireNamespace("gtrellis", quietly = TRUE)) {
install_github("jokergoo/gtrellis")
}
if (!requireNamespace("tidyverse", quietly = TRUE)) {
install.packages("tidyverse")
}
if (!requireNamespace("ggplotify", quietly = TRUE)) {
install.packages("ggplotify")
}
if (!requireNamespace("RColorBrewer", quietly = TRUE)) {
install.packages("RColorBrewer")
}
# 加载包
library(circlize)
library(ComplexHeatmap)
library(gtrellis)
library(tidyverse)
library(ggplotify)
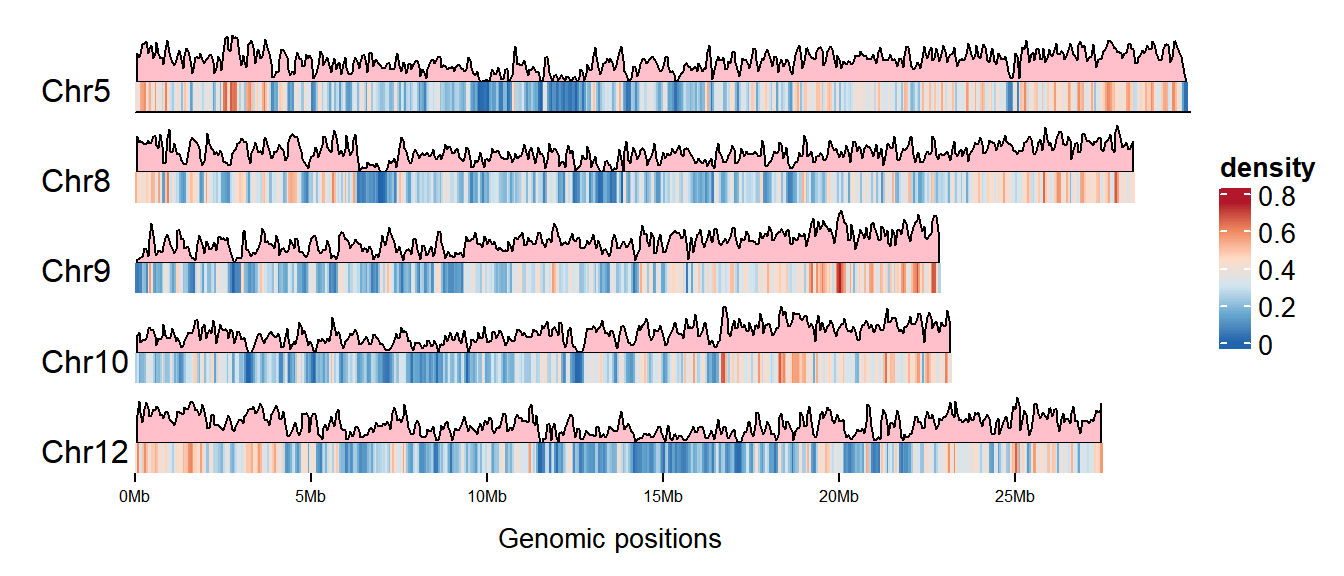
library(RColorBrewer)基因密度图
注记
Hiplot 网站
本页面为 Hiplot Gene Density 插件的源码版本教程,您也可以使用 Hiplot 网站实现无代码绘图,更多信息请查看以下链接:
染色体数据展示。
环境配置
系统: Cross-platform (Linux/MacOS/Windows)
编程语言: R
依赖包:
circlize;ComplexHeatmap;gtrellis;tidyverse;ggplotify;RColorBrewer
数据准备
# 加载数据
data1 <- read.delim("files/Hiplot/060-gene-density-data1.txt", header = T)
data2 <- read.delim("files/Hiplot/060-gene-density-data2.txt", header = T)
# 整理数据格式
chrNum <- str_replace(unique(data1$chr), "Chr|chr", "")
data1$chr <- factor(data1$chr, levels = paste0("Chr", chrNum))
data2$chr <- factor(data2$chr, levels = paste0("Chr", chrNum))
# 设置窗口计算基因密度
windows <- 100 * 1000 # 默认:100kb
gene_density <- genomicDensity(data2, window.size = windows)
gene_density$chr <- factor(gene_density$chr,
levels = paste0("Chr", chrNum)
)
# 查看数据
head(data1) chr start end
1 Chr5 0 29958434
2 Chr8 0 28443022
3 Chr9 0 23012720
4 Chr10 0 23207287
5 Chr12 0 27531856head(data2) chr start end
1 Chr10 38648 40060
2 Chr10 45941 58338
3 Chr10 67119 72971
4 Chr10 75410 76305
5 Chr10 80964 82250
6 Chr10 94798 97746可视化
# 设置画板颜色
palettes <- c("#B2182B","#EF8A62","#FDDBC7","#D1E5F0","#67A9CF","#2166AC")
col_fun <- colorRamp2(
seq(0, max(gene_density[[4]]), length = 6), rev(palettes)
)
cm <- ColorMapping(col_fun = col_fun)
# 设置图例
lgd <- color_mapping_legend(
cm, plot = F, title = "density", color_bar = "continuous"
)
# 绘制基因密度分布热图
p <- as.ggplot(function() {
gtrellis_layout(
data1, n_track = 2, ncol = 1, byrow = FALSE,
track_axis = FALSE, add_name_track = FALSE,
xpadding = c(0.1, 0), gap = unit(1, "mm"),
track_height = unit.c(unit(1, "null"), unit(4, "mm")),
track_ylim = c(0, max(gene_density[[4]]), 0, 1),
border = FALSE, asist_ticks = FALSE,
legend = lgd
)
# 添加基因面积图 track
add_lines_track(gene_density, gene_density[[4]],
area = TRUE, gp = gpar(fill = "pink"))
# 添加基因密度热图 track
add_heatmap_track(gene_density, gene_density[[4]], fill = col_fun)
add_track(track = 2, clip = FALSE, panel_fun = function(gr) {
chr <- get_cell_meta_data("name")
if (chr == paste("Chr", length(chrNum), sep = "")) {
grid.lines(get_cell_meta_data("xlim"), unit(c(0, 0), "npc"),
default.units = "native")
}
grid.text(chr, x = 0.01, y = 0.38, just = c("left", "bottom"))
})
circos.clear()
})
p