# Install packages
if (!requireNamespace("umap", quietly = TRUE)) {
install.packages("umap")
}
if (!requireNamespace("ggpubr", quietly = TRUE)) {
install.packages("ggpubr")
}
# Load packages
library(umap)
library(ggpubr)UMAP
Note
Hiplot website
This page is the tutorial for source code version of the Hiplot UMAP plugin. You can also use the Hiplot website to achieve no code ploting. For more information please see the following link:
UMAP is a nonlinear dimensionality reduction algorithm suitable for high-dimensional data reduction to two or three dimensions and visualization. The algorithm can make the t distribution of points with greater similarity closer in the lower dimensional space. For low similarity points, the t distribution is farther away in the low dimensional space.
Setup
System Requirements: Cross-platform (Linux/MacOS/Windows)
Programming language: R
Dependent packages:
umap;ggpubr
Data Preparation
The loaded data are the data set (gene name and corresponding gene expression value) and sample information (sample name and grouping).
# Load data
data1 <- read.delim("files/Hiplot/176-umap-data1.txt", header = T)
data2 <- read.delim("files/Hiplot/176-umap-data2.txt", header = T)
# convert data structure
sample.info <- data2
rownames(data1) <- data1[, 1]
data1 <- as.matrix(data1[, -1])
## umap
set.seed(123)
umap_info <- umap(t(data1))
colnames(umap_info$layout) <- c("UMAP_1", "UMAP_2")
# handle data
umap_data <- data.frame(
sample = colnames(data1),
umap_info$layout
)
colorBy <- sample.info[match(colnames(data1), sample.info[, 1]), "Species"]
colorBy <- factor(colorBy, level = colorBy[!duplicated(colorBy)])
umap_data$colorBy = colorBy
shapeBy <- NULL
# View data
head(data1[,1:5]) M1 M2 M3 M4 M5
Sepal.Length 5.1 4.9 4.7 4.6 5.0
Sepal.Width 3.5 3.0 3.2 3.1 3.6
Petal.Length 1.4 1.4 1.3 1.5 1.4
Petal.Width 0.2 0.2 0.2 0.2 0.2head(data2) Samples Species
1 M1 setosa
2 M2 setosa
3 M3 setosa
4 M4 setosa
5 M5 setosa
6 M6 setosaVisualization
# umap
p <- ggscatter(data = umap_data, x = "UMAP_1", y = "UMAP_2", size = 2,
palette = "lancet", color = "colorBy") +
labs(color = "group") +
ggtitle("UMAP Plot") +
theme_classic() +
theme(text = element_text(family = "Arial"),
plot.title = element_text(size = 12,hjust = 0.5),
axis.title = element_text(size = 12),
axis.text = element_text(size = 10),
axis.text.x = element_text(angle = 0, hjust = 0.5,vjust = 1),
legend.position = "right",
legend.direction = "vertical",
legend.title = element_text(size = 10),
legend.text = element_text(size = 10))
p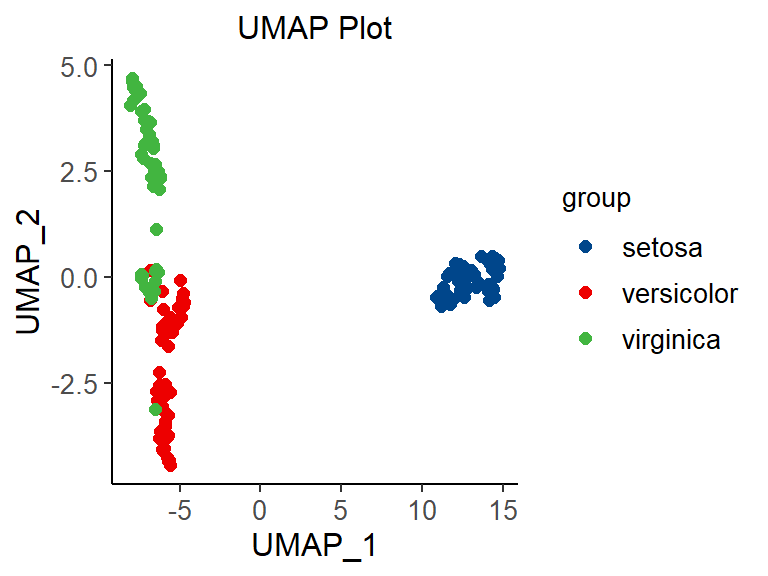
Different colors represent different samples, which is the same as PCA (principal component analysis) graphic interpretation. The difference lies in the visualization effect. For dissimilar points in T-SNE, a small distance will generate a large gradient to repel them.